
Published online: 14.01.2020
Author: Kath Rothwell
In my opinion, the reason that Mössbauer spectroscopy is such a powerful analytical tool for earth and environmental sciences is because of its isotope specificity.
The reason for this specificity is due to the Mössbauer Effect itself. For 57Fe - Mössbauer, we use the radioactive isotope 57Co as a source, which decays to 57Fe through the emission of a gamma photon. In principal, if no energy is lost in this process, the emitted gamma photons could be absorbed by other identical nuclei, i.e. exclusively 57Fe. Unfortunately, no suitable parent isotopes exist to allow Mössbauer spectroscopy to work with other Fe isotopes. However, due to the law of conservation of momentum, in reality both the decaying and absorbing nuclei will recoil as they emit or absorb a photon, thus slightly altering the energy of the photon rendering resonant absorption improbable (see figure 1).

Nonetheless, the Mössbauer Effect describes the recoil-free resonant emission and absorption of gamma photons, which is possible due to energy-level changes in the vibrational modes of a solid lattice, which counteract the energy lost through nuclear recoil (Figure 2).

For more detailed information about the underlying physics Dyar et al., (2006) is a very useful article.
As Mössbauer spectroscopy is specific to 57Fe, other Fe isotopes are invisible to Mössbauer. At first, this may seem like a hindrance, however, it is actually an extremely useful feature when combined with the use of stable Fe isotopes.
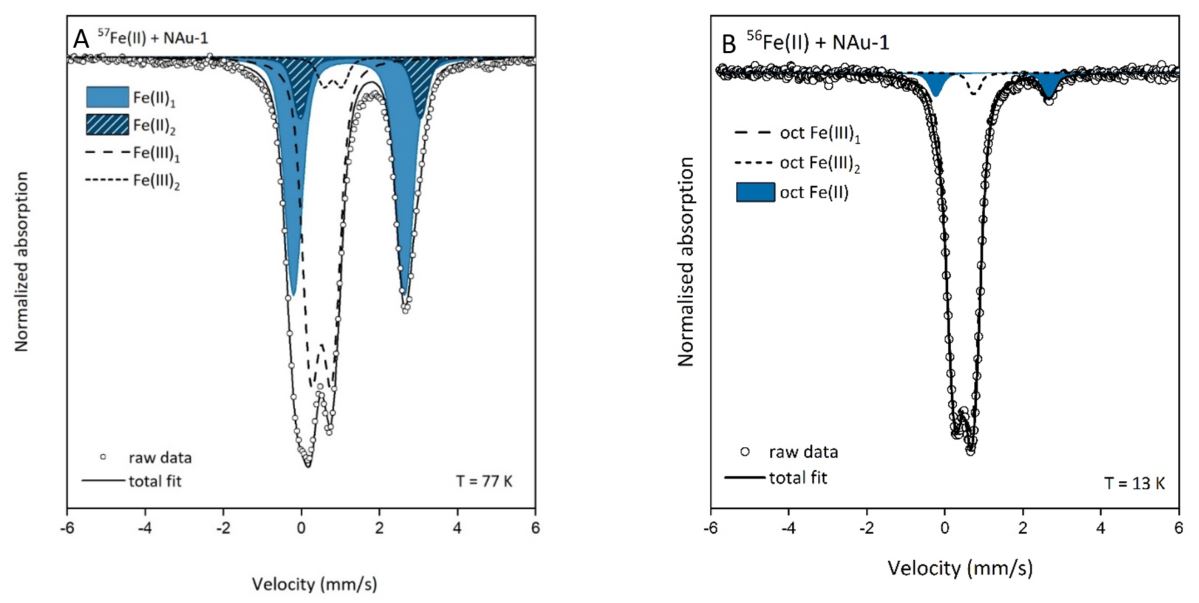
In my research, I use stable Fe isotopes to probe either changes in the structure of Fe-bearing clay minerals that have been reacted with aqueous Fe2+ or the formation of new Fe-bearing phases that precipitate as a result. I start with either a synthetic Fe-free or 56Fe-bearing mineral (Panel A below) or a naturally occurring Fe-bearing mineral (Panel B below). For an Fe-free or exclusively 56Fe-bearing phase no Mössbauer signal is visible and for a natural Fe-containing phase the signal for only the naturally abundant structural Fe is seen.

If I aim to characterise newly formed Fe-containing precipitates, I react my initial mineral with enriched aqueous 57Fe2+ prior to Mössbauer analysis (Figure 4, Panel A below). If I started with an Fe-free or solely 56Fe-containing phase then the spectra will exclusively show the precipitate. However, if I started with a mineral containing some natural abundance Fe (of which an average 2.119% comprises 57Fe) then this will also be visible in the spectra, although due to the enrichment factor of the 57Fe this is usually only a very small amount of the spectral area (< 0.1 % in the example shown in Panel A). Of course, if you start with an Fe-free or exclusively 56Fe-containing sample then natural abundance aqueous Fe could also be used to characterise a surface precipitate but the use of 57Fe allows for more rapid spectral acquisitions.
On the other hand, if I am interested in what changes occur in the structure of a Fe-bearing mineral after reaction with aqueous Fe then I react my sample with enriched 56Fe2+ (Figure 4, Panel B below). In this case, only the changes that occur in the naturally abundant Fe in the mineral structure are visible in the spectra and for the example in Panel B we can confirm that reduction of structural Fe has occurred by the appearance of the blue doublet that is absent in the starting material.
I study the transformation of Fe-bearing clay minerals and the characterisation of precipitated phases as they are important for the reductive transformation of a range of environmental contaminants including chlorinated solvents, radionuclides, potentially toxic metal(loid)s and several others, and the use of these isotope games has been widely used in environmental science to study reactive iron minerals. For example, Williams et al., (2004) used this approach to show that Fe atom exchange and recrystallisation occurs between Fe oxides and aqueous Fe2+, which is extremely interesting as iron oxides are a major sink of potentially toxic trace elements that may be mobilised upon recrystallization. They used a synthetic goethite containing only 56Fe and Mössbauer analysis of the synthetic goethite only showed background noise as no Mössbauer-active 57Fe was present, although goethite was confirmed using XRD. They reacted the 56Fe-goethite with 57Fe2+ for 6 hours and repeated the Mössbauer measurement, which showed a spectrum containing a sextet characteristic of goethite thus showing that the 57Fe2+ had not just sorbed to the goethite surface but had facilitated atom exchange between the solid and aqueous Fe pools.
Following on from Williams et al's work, several others have used this approach to show that Fe2+ catalysed transformation of Fe oxides occurs in natural samples and soils. Therefore, whether at lab or field scales the use of stable Fe isotopes allows Mössbauer spectroscopy to not only characterise Fe minerals but also to study mineral transformation and to quantify changes in and between associated iron pools (for example, clay mineral Fe and precipitated Fe).
Thanks to Craig Thompson for assistance with the figures depicting recoil and resonant absorption
Did you like this tutorial? Is there anything which can be improved? Or perhaps you want to contribute you own tutorial? If so, please get in touch.